


Therapeutics around the central dogma: DNA
A study note on gene therapy and gene editing landscape: MoA and delivery


Biodraft is a biotech blog started by Liang Chang (PhD student at Broad/Harvard) and Kirill Karlin, MD (pathology resident at BIDMC/HMS). As trainees in science/medicine and students in biotech, we post our monthly biotech study notes here, which are mostly deep-dive analyses of interesting biotech areas we recently learned. Click here for a introduction article about Biodraft, Liang, and Kirill.
This is the first article for my “therapeutics around the central dogma” study note series. Recent exciting news about gene therapy and gene editing (especially the clinical proof-of-concept data of in vivo CRISPR gene editing in ATTR patients by Intellia) inspired me to systematically learn about the technologies and players in this space. This project soon became larger than expected, as I realized that for many diseases there are many modalities (and all the pros and cons of each) aiming to solve the problem. I decided to construct a framework for “therapeutics around the central dogma”- the MoA is to modulate specific DNAs/RNAs/proteins. I’m trying to post my study note along the way since writing is an effective way to internalize my learning. This series will likely have 4 articles: DNA, RNA, protein, and disease deep dive (tentatively on sickle cell disease and ATTR).
Without further ado, here’s my 1st study note on targeting DNAs!
The long-term or even permanent modulation of DNA, the genetic blueprint in each and every cell in humans, has been an attractive therapeutic concept for many diseases. For single-gene diseases (mostly inherited rare diseases) where the root cause is on the DNA level, adding a normal copy of functional DNA or editing the endogenous copies brings hope to a single-dose cure. For many chronic diseases that require daily medication -inconveniences for patients and compliance headaches for physicians, targeting DNAs can potentially change the picture for disease management. For complicated diseases like cancer and autoimmune diseases, ex-vivo genetic engineering of cells can “teach old dog new tricks” and bring great hopes for the next disease-modifying breakthrough.
The past few years have been truly exciting for DNA-targeting therapeutics. On the technology front, the discovery of CRISPR revolutionized gene editing, and the rich and versatile Cas protein toolkit opens many new doors for therapeutic application. On the delivery front, innovations in AAV optimization and emerging non-viral approaches have made solid progress from labs to patients. Yet many challenges, especially in vivo delivery efficacy and safety, leave much open space for ongoing and future efforts in both academia and industry.
Technology/Mechanism of Action (MoA)
Technologies/MoAs for DNA therapeutic application can be categorized into 3 bins: Addition (adding a copy of normal/functional DNA), Removal (remove unwanted DNA), and Modification (editing/replacing unwanted DNA). Below is a map for these technologies, and the corresponding delivery methods.
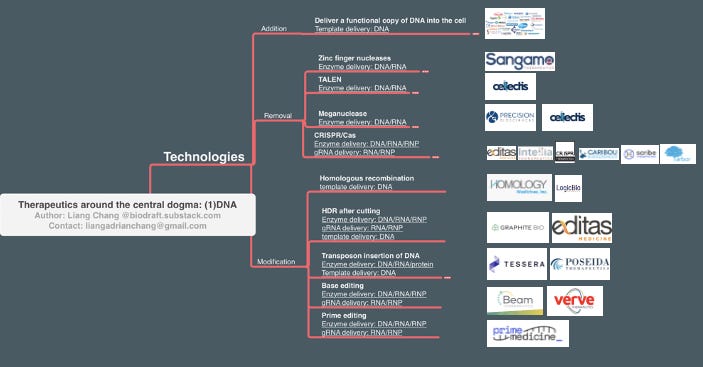
Addition (gene therapy)
To treat diseases caused by the loss-of-function of genes such as spinal muscle atrophy (SMA, loss of functional SMN1) or hemophilia A (loss of Factor VIII), or develop therapeutics by introducing a new piece of the gene (such as CAR-T), the most intuitive way is to deliver a functional copy DNA into the cell. Optimal DNA delivery is the most important consideration for all DNA targeting applications, and I will go through this space in detail in the delivery section. The death of Jesse Gelsinger in a 1999 gene therapy clinical trial using an adenovirus vector halted this field for a decade, but recent progress in alternative delivery methods brings a lot of excitement and hope. In a nutshell, for ex vivo applications there are several tools to choose from, largely depending on cell type and manufacturing considerations. For in vivo applications, the adeno-associated virus (AAV) vector is the most popular choice given its mild immunogenicity and episomal delivery feature, and many companies, large and small, are actively working on AAV vectors (although some recent data indicate that the immunogenicity and DNA integration carcinogenesis of AAV cannot be ignored. I will expand in the delivery section).
In addition to the delivery vector, there are several additional considerations for effective DNA/gene delivery into the cell. Some genes are very large and can exceed the upper limit of delivery vectors. For example, the DNA packaging limit of AAV vectors is <5kb, but the full-length cDNA of the human FVIII gene (for hemophilia A) is ~6kb and of the Dystrophin gene (for DMD) is >20kb. Careful engineering of a truncated but functional “mini-gene”, or less ideally splitting a large gene and package each segment separately, need some rational designs and trial-and-error in the lab. Also, protomer selection is not always trivial. While a strong, ubiquitous promotor can maximize the chance for successful gene expression, alternative promoters of different tissue/cell type selectivity and expression strength can be another differentiation factor to optimize the therapeutic index.
Other methods, using certain repeating elements in the genome for site-specific “cut and paste”, can also be used to insert a piece of DNA into the specific locations in the genome. These methods, including homologous recombination, homology-directed repair, and transposon, will be covered in the “modification” section.
Removal (gene editing)
Here’s the shared feature of current technologies that remove a functional gene on the DNA level: use a nuclease to create a precise DNA double-strand break at specific locations in the genome (ideally at the early position or in the Pfam domain of the gene of interest, GOI), let the cell’s error-prone NHEJ mechanism to fix the double-strand break and leave small insertions or deletions (indels) around the cutting site, and these indels can disrupt the reading frame of GOI and create premature stop codon, leading to loss of GOI.

Finding a robust programmable site-specific DNA cutter is a key requirement for precision gene removal in the genome. In the pre-CRISPR era, zinc finger nucleases (ZFNs), TALENs, and meganucleases represented the older generation of gene editing tools that have been translated into the clinic. Cellectis and Precision BioSciences had a messy 5-year patent fight over meganuclease and finally reached a settlement in 2014, while Cellectis shifted focus to develop TALENs. Both companies currently work on off-the-shelf CAR-T products, and both have anti-CD19 candidates currently in Phase 1 clinical trials. Sangamo Therapeutics, a biotech company working on ZFNs since 1995, is a good example to show the long and winding road from exciting technology to patient-benefiting therapeutics. After pivoting from terminated clinical programs in diabetic neuropathy, HIV, and MPS II, and recovering from walk-aways from several pharma partners, Sangamo now has a hemophilia A program currently in phase 3 in partnership with Pfizer. One common drawback about these pre-CRISPR gene-editing technologies is that they rely on protein permutations to recognize a specific segment of DNA. Thus, for each new targeting site, a new round of protein engineering (or mix&match) is required, which can be slow and cumbersome.

The discovery of CRISPR completely changed the field of gene editing. Now instead of using protein combinations to recognize the DNA-targeting sites, a short segment of unique guide RNA is used without the need to change the Cas protein. Since small RNA is much easier to design and synthesize, the flexibility of gene editing opens massive opportunities for new therapeutic applications.

Here’s a table summarizing companies working on CRISPR/Cas gene editing. Editas, Intellia, CRISPR are the first batch of companies founded by the CRISPR pioneers (Doudna, Charpentier, Zhang, Church). A few years down the line, there are many similarities among these companies: all went public with high valuations hard to be explained by DCF models, all had candidates in the clinic, all secured pharma partnerships, and all working on ex vivo applications including SCD and cell therapies. Nonetheless, different therapeutic focuses have clearly emerged. Editas, the first mover in the game, chose a safer route to establish a proof-of-principle in vivo gene editing by working on AAV delivery to treat genetic eye diseases. Gene therapy for ocular diseases just requires a low amount localized injection of virus vector and eyes are immune privileged, favorable for safety and manufacturing considerations. Spark Therapeutics (acquired by Roche), one of the first players in the AAV space, secured the first FDA approval of AAV-vectored gene therapy in eye disease in 2017. CRISPR Therapeutics is the fastest mover in hematopoietic diseases that can be fixed by ex vivo editing- sickle cell disease (SCD) and beta-thalassemia. Earlier this year they reported a positive Phase 1 readout, showing 80% editing efficiency and > 1-year clinical benefit in patients. Intellia Therapeutics takes a riskier attempt at in vivo gene editing. They used lipid-nanoparticle (LNP) delivery of Cas9 mRNA together with gRNA to patients through systematic delivery (IV), aiming to knock out mutated TTR gene in patients’ liver, reduce the production of misfolded TTR protein, and fixed the root cause of hAATR. They recently reported positive PD data of the Phase 1 trial, showing a >80% serum TTR reduction in the highest 0.3mh/kg group patients. This is a major milestone in gene editing therapeutics that CRISPR can be delivered through systematic delivery and works in humans.

Caribou Biosciences, another CRISPR company backed by Doudna, also recently went public. It solely focused on ex vivo editing to make allogeneic CAR-T and CAR-NK cells. There are some interesting stories between Caribou and Intellia, since Caribou co-founded and licensed the Doudna CRISPR/Cas9 patent to Intellia, but later got into some patent disputes. I’m not entirely sure whether Caribou’s emphasis on hybrid DNA-RNA guides and Cas12a is related to the IP situation. Another recent Doudna lab spinoff, Scribe Biosciences, focused on the commercialization of CasX (Cas12e). On the east coast, Arbor Biotechnologies is backed by Feng Zhang and working on new CRISPR transposases and new nucleases. Their pipeline, though super early, seems to focus on in vivo editing of liver diseases. They recently secured a collaboration deal with Vertex.
Modification
Modifying the genome -replacing the faulty DNA base pairs/segments or inserting a piece of DNA to specific genomic locations, is more challenging than DNA addition or removal. But fortunately, mother nature leaves us with some mechanisms to modify DNA as starting points for engineering campaigns.
Homologous recombination is a DNA repair mechanism that uses the 2nd copy of DNA as a template. The DNA segments flanking both the 5’ and 3’ end of the repairing site, “homology arms”, can be hijacked by delivering a target gene with the ideal homology arms. This process also occurs during meiosis to increase the genetic diversity of the germ cells. In somatic cells without DNA breaks, however, the frequency of homologous recombination is low, and a selection marker is needed to select cells with the expected recombination. Homologous Medicine and Logic Therapeutics are two current players that use homologous recombinations, and their therapeutics applications are mostly limited to ex vivo editing. Graphite Bio, on the other hand, uses Cas9 to cut at targeting loci to trigger the DNA repair machinery, and uses AAV to deliver a gene template for homology-directed repair. They also start with ex vivo editing, joining the competitive race along with other gene-editing companies to cure sickle cell disease.

Transposons are the jumping element that viruses left in our genome after many years of attack, defense, and co-evolution. These transposons can be used for transposases to “cut-and-paste” DNA of interest at specific locations in the genome. Nearly 30 years after Barbara McClintock was awarded Nobel for the transposon discovery, there is still a lot to learn about these jumping elements in the genome. Sleeping Beauty and piggyBac are the two most-adapted transposon systems for molecular biology applications, and Poseida Therapeutics uses piggyBac to make allogeneic CAR-T cells. Tessera Therapeutics, started by Flagship Pioneering in 2018, has a much bigger ambition. They aim to use transposons as a “gene writing” platform. While they haven’t disclosed their pipeline, the recent $230M fundraising and adding former Beigene CFO Howard Liang to the roster indicate that they are ready for future big stages.

The engineering of CRISPR-Cas beyond DNA cutting also brings new promises for gene modification tools. Beam Therapeutics, a spin-off from David Liu’s lab, working on therapeutics applications of “beam editors” -deaminase linked with dCas9 protein. In this case, Cas9 + gRNA serves as a precise genome localization tool, and the deaminase does the job to change nucleotides at certain locations. Prime Medicine, another recent David Liu lab spinoff, is based on a 2019 technology that fusing Cas9 nickase with reverse transcriptase. The resulting “prime-editor” can generate a lot of DNA repair templates of interest in situ of the DNA cutting site, increasing the chance to get successful HDR, and therefore making a variety kind of DNA modification possible beyond single-base-pair editing.
Delivery
From a functional piece of cDNA all the way to the complicated prime editor, finding the right tool to deliver DNA into the right cells is critical. Here’s a map I made to cover many DNA delivery technologies, what’s possible to achieve by different technologies (clinical-stage indications), and some of the players.

Ex vivo
Comparing with delivering DNAs directly into humans, starting with isolated cells outside of the body is a lot more straightforward to do. Some high-efficiency tools like electroporation are possible pretty much only in a lab (Still, companies like Inovio and Eyevensys have developed devices to directly electroporate DNAs to muscle and eye), and selections markers can be used to enrich cells needed.

From the delivery technology front, electroporation and lentiviral/retroviral vectors and are most commonly used. Electroporation can be used not only to deliver DNA, but also ribonucleoprotein (RNP, a complex of Cas protein and gRNA) for CRISPR gene editing applications. It’s easier to use without worrying about issues related to virus vectors, but scaling up the manufacturing process can be problematic. Lentiviral/retroviral vector directly inserts DNA into the genome of the infected cell, allowing high efficiency and durable delivery in many cell types. However, given the random integration nature of lentivirus and retrovirus, insertional mutagenesis is always a risk to consider. For retrovirus vectors, two clinical trials in X-SCID patients led to restored immune function in the majority of patients, but 5/20 patients developed T-cell leukemia caused by virus vector integration around the LMO2 gene. For lentiviral vectors, AML and MDS cases in patients treated with Bluebird Bio’s LentiGlobin raised big safety concerns, although the conditioning drug busulfan can also be the reason behind these cancer cases. Alternative ex vivo DNA delivery methods also draw a lot of attention in recent years. SQZ Biotech, a Robert Langer lab spinoff company, uses a microfluid device with capillaries smaller than cell diameters toliterally “squeeze” DNAs into the cells. Sana Biotechnology, another Flagship company focusing on iPSC-derived cell therapies, uses “fusosome” - particles containing virus-derived fusogen proteins, to deliver DNAs into the cell ( I have to dig into the company S1 to find this information).
Immune cells for oncology indications are the battlefield for many big and small companies. After the approval of autologous anti-CD19 and anti-BCMA CAR-T therapies, many companies include Allogene Therapeutics, Precision Biosciences, and Caribou Biosciences seek to develop “off-the-shelf” allogeneic CAR-Ts, first against these two well-derisked targets. In addition to CAR-T, CAR-NK (Nkarta, Fate, Takeda, etc), CAR-γδT (Adicet, Eureka, Gadeta, etc), and CAR-Macrophage (Carisma) are among other “CAR-family” effector immune cells that are engineered to tackle cancer. SQZ Biotech works on “squeezed” antigen-presenting cells (APCs) to boost immune response for cancer, and also recently joined the race with Rubius Therapeutics to develop genetic-engineered red blood cells (RBCs) for both oncology and autoimmune disease applications. iPSC cells, which can be potentially developed into many different lineages, also recently attracted a lot of attention as the starting point for genetic engineering. Sana Biotechnology and Century Therapeutics work on iPSC-derived CAR-T and CAR-NK cells, whereas CRISPR Therapeutics in collaboration with Viacyte has an interesting early program in diabetes.
Ex vivo gene editing and gene therapy with hematopoietic stem cells also bring hope to many patients with rare genetic diseases (Side note: I recently started to read about rare diseases and was deeply impressed by decades of effort by patients, parents, scientists, and biotech companies to make therapeutics from hope to reality. It definitely worths a future study note on rare diseases and the early stories of Genzyme/Biomarin/Vertex). For many of these diseases, stem cell transplantation is a risky bet but not always possible, but the editing of a patient’s own HSC and transfusion back solves many problems of a transplant. For sickle cell disease and beta-thalassemia, there are many orthogonal technologies currently in clinical trials, including lentiviral gene overexpression (Bluebird), CRISPR (CRISPR, Intellia), base-editing (Beam), and CRISPR HDR (Graphite). It will take some time to see which technology is superior, or if multiple tricks can get the same job done. For diseases in which the primary deficit is not from the hematopoietic lineage, such as lysosomal storage disorders, Bluebird Bio, Orchard Therapeutics, Avrobio, among other companies, are also trying to ex vivo fix the HSCs given that stem cell transplantation can bring clinical benefit. In vivo gene editing might offer better solutions, as I will discuss in the next section.
In vivo

The insertional mutagenesis risk of lentivirus and retrovirus makes them less ideal to deliver directly in humans. Viruses that deliver episomal DNAs (rather than integrating DNAs into the genome) are therefore safer and more attractive for in vivo DNA delivery applications. Adenovirus vector is the first being tried, but unfortunately leading to Jesse Gelsinger’s death due to fatal liver inflammation. The immunogenicity of adenovirus vectors is a curse for gene therapy applications, but a blessing for vaccine development. Several COVID vaccines, including JnJ’s, AstraZeneca’s, Cansino’s, and Sputnik all use adenovirus vector to deliver the SARS-CoV-2 spike protein DNA sequence to generate a robust immune response. Herpes simplex virus (HSV) is another virus that delivers episomal DNA. They are mostly used to be engineered as oncolytic virus, selectively replicating in and killing proliferative cancer cells, provoking immune response at the same time. Krystal Biotech takes a different approach to use HSV for dermatology and respiratory gene therapy delivery. Their lead candidate is in a phase 3 trial treating Dystrophic EB.
Adeno-associated virus (AAV) vector is the brightest star in the gene therapy universe. AAV vector is an ideal DNA delivery vehicle with the right combination of low immunogenicity and episomal feature. A comprehensive AAV landscape review is beyond the scope of this article, and I’ll mostly let my landscape map speak for itself. With AAV localized injection, eye/ocular and central nervous system (CNS) are the most popular therapeutic areas. As mentioned earlier in the article, the eye is the prime organ for gene therapy and gene editing, bringing hope to patients with genetic diseases affecting the eye. Directly deliver AAV to the CNS system is another attractive direction, given that many neurological genetic diseases such as Huntington’s have huge unmet needs. With systematic delivery, the landmark approval of Zolgensma to treat SMA demonstrated that IP injection of AAV9 can successfully deliver to CNS. IV AAV delivery to target the liver also established clinical proof-of-concept, and several companies have phase 3 candidates for both hemophilia A and B. IV AAV delivery to the muscle cells, a seemingly very difficult task, will get its ultimate test of phase 3 trials in DMD.

Beyond the broad enthusiasm in the AAV vector field, some problems remain to be solved. (1) AAV gene delivery efficacy in a longer time horizon needs to be proven, especially given that AAV is regarded (and is priced) as a long-term cure for diseases. But a sneak peek of that from Biomarin’s 5-year hemophilia A data looks not quite promising, as patients had a median of 8.2% the amount of factor VIII levels to a healthy individual vs 60.3% after a one-year follow-up; (2) re-dosing a patient with the same serotype of AAV vector is currently not possible, given that the immune system will recognize and eliminate the vector at the second entry to the body. Similarly, if patients had prior exposure to certain AAV serotypes, they will not benefit from the treatment; (3) the safety issues of AAV vectors gradually raised concerns. In an XLMTM trial run by Audentes Therapeutics, 3 patients died in the high dose group with some potential links to liver dysfunction, which indicates that the immunogenicity of AAV cannot be ignored especially with high doses. In addition, FDA put a clinical hold on Biomarin’s PKU gene therapy candidate BMN307 because in a preclinical mouse study, 6/7 animals in the highest dose group (2e14 Vg/kg) had liver adenomas with evidence of AAV genomic integration. Thus, continuous innovation in the AAV gene therapy field both from the industry (eg. George Church lab spinoff Dyno Therapeutics uses deep learning to facilitate AAV capsid engineering ) and academia (eg. this recent Nature article uses a gene-splicing drug as an “on switch” for gene therapy) are needed now more than ever to continuously push the boundaries for therapeutics forward.
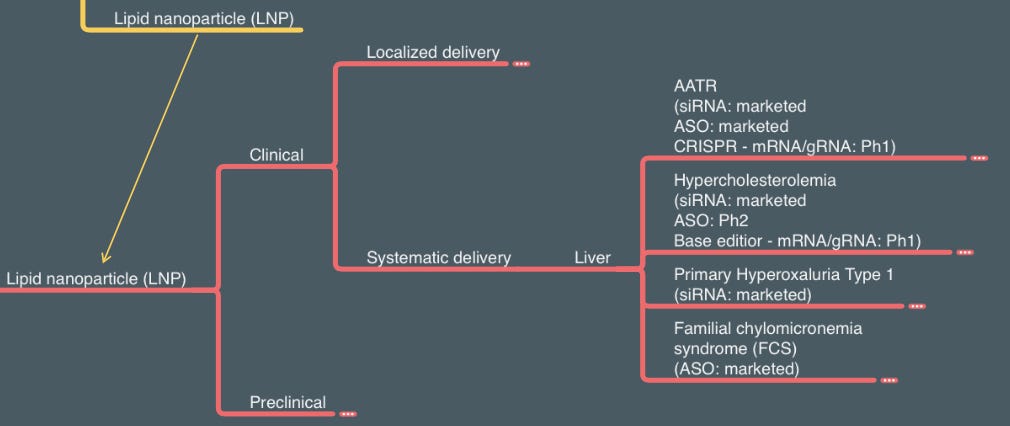
Lipid nanoparticles (LNP) can also be used to deliver DNAs to the cell, and Generation Bio is one of the few companies working in this space. However, the true power of LNP is to deliver mRNA, both locally and systematically. I borrowed the systematical delivery section of LNP from the RNA delivery section (stay tuned for my future RNA technology + delivery article!) since several gene-editing technologies reply on LNP delivery of Cas protein mRNA and gRNA to the liver. Gene editing can provide a “once and done” solution to both genetic diseases (AATR as the first indication for Intellia’s CRISPR knockout) and chronic disease (inherited hypercholesterolemia as the first indication for Verve’s base editor). It will be exciting to see more clinical data in the next few years.
Future
This is the best time for DNA therapeutics. Advances in technologies (especially after the CRISPR revolution) and delivery (especially around AAV) enabled exciting companies and therapeutics pipelines spanning across multiple fields -gene therapy, gene editing, cell therapy, genetic diseases, oncology, cardiovascular diseases, and beyond. This is also an uncertain time for DNA therapeutics. Drug discovery is risky, and it’s certainly not an easy job to translate first-of-its-kind technologies to first-in-class medicine. I have some questions to guide myself into the future world with DNA therapeutics:
Technology
Do we have the right tools (Prime editing/transposons/etc) to modify DNAs beyond the single base-pair level?
For genetic diseases that can be addressed by multiple therapeutic modalities (eg. hATTR, small molecule stabilizer vs. RNAi vs. CRISPR), what are the pros and cons for each strategy, and how would that translate to physicians’ decisions?
In addition to well-characterized single genetic diseases, can we slice some bigger, more heterogeneous diseases and identify patients that can benefit from current technologies? Can we apply DNA therapeutics to treat chronic diseases?
What’s the long-term safety for these somewhat irreversible therapies?
Delivery
Would the chance of genomic integration and cancer risk be AAV’s Achilles heel?
How can we further improve the current limitations of AAVs (immunogenicity, re-dosing, efficacy duration)? Can we find or engineer new AAV capsids with improved features (organ selectivity, high titers in manufacturing, etc)?
What delivery efficiency and durability could non-viral delivery methods achieve? How would lipid + mRNA/circular DNA compete with AAV?
With systematic delivery, which organs (beyond the liver) can be eventually reached at sufficient efficiency to translate into clinical benefit?
For ex vivo cell therapies, especially allogeneic immune cells like CAR-T or CAR-NK, would they reach a similar efficacy ballpark to autologous CAR-T? Can we see clinical benefits beyond CD19 and BCMA, especially in solid tumors?
Thanks for stay with me to the end! If you like it, please subscribe to Biodraft for future articles from Kirill and myself. Feel free to contact me at liangadrianchang AT gmail.com. See you next time!
About the author

I’m Liang, a 5th year PhD student at the Broad Institute and Harvard Medical School I work on new functional genomic technologies to find cancer therapeutic targets and mechanisms (here’s my first-author Cancer Cell perspective article on targeting pan-essential genes). I’m also a senior business development at Harvard OTD, helping to commercialize early life science technologies right out of Harvard labs. I’m excited about learning the science and business around new therapeutics, from lab to patients. Contact me: Twitter, Linkedin
I love this!! Don’t mean to spam you, but this paper has inspired me so much and it shows nicely where the future of AAV has headed and i have to share it with you😊 https://www.nature.com/articles/s41593-020-0692-9